全基因組測序(WGS)是下一代測序技術,用于快速,低成本地確定生物體的完整基因組序列。基因組的深度測序對于臨床研究的意義重大,解讀WGS數據并了解基因組突變在健康和疾病中的重要性是精準醫療的基石。
WGS分析流程能分為三大塊,數據處理、檢測變異和綜合分析,具體如下圖所示:
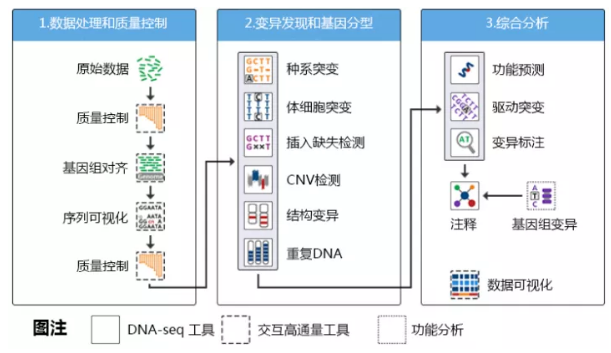
由于WGS現在已經非常成熟了,因此這里面的每一步驟都能在網上找到相應的工具。
質量控制
在高通量測序中,有時候會出現低質量讀數和污染讀數等問題,會影響接下去的分析結果。因此,質量控制(QC)對于原始NGS數據至關重要。現在質量控制軟件主要用的是 NGS-QC Generator,它可以從與特定NGS概況相關聯排序讀數的分布推斷出質量指標。
基因組對齊
高通量測序分析中需要將生成的讀數對準(映射)到參考序列。此時,就需要用到Bowtie和BWA。前者能以每小時超過2500萬個35 bp讀數的速率將短DNA序列(讀取)與人類基因組進行比對。后者針對大型參考基因組(如人類基因組)繪制低分歧序列。
序列可視化
可視化讀取對齊是使用現有數據驗證候選結構變體(SV)的最有效方式。這可以用IGV來完成。高性能的查看器可以有效地處理大型異構數據集,同時在所有基因組分辨率級別提供直觀的用戶體驗。IGV的一個關鍵特征是其關注于基因組研究的綜合性質,支持基于陣列和下一代測序數據,以及臨床和表型數據的整合。

變異檢測
都是各種基因變異的檢測手段,就放一起說了。種系突變、體細胞突變和插入缺失檢測這三個的檢測都可以用一個軟件,SAMtools。這是用于與高通量排序數據進行交互的程序。它可以處理SAM / BAM / CRAM格式的讀取,寫入,編輯,索引,查看和轉換SAM / BAM / CRAM格式。SAMtools還可以索引FASTA格式的引用序列或從索引引用序列中提取子序列。

拷貝數變異(CNV)是遺傳變異的常見來源,涉及許多基因組障礙。CNV是基因組中結構變異(SV)的一種形式。通常,CNV是指大于1kbp的DNA片段的重復或缺失。CNV檢測可以用CNVnator,它可以從家族和群體基因組測序中發現基因型、表征典型和非典型CNV的方法。CNVnator具有靈敏度高(86%-96%),假陽性低(3%-20%),基因分型準確率高(93%-95%)的特點。
短串聯重復序列(STR)是DNA重復的一種,重復單位為2-6bp,重復次數10~60多次,基因片段,400bp以下。STR是存在于人類基因組DNA中的一類具有長度多態性的DNA序列,不同數目的核心序列呈串聯重復排列,而呈現出長度多態性,通常多態性片段長度在100-300bp。
STR的長度變異性與許多物種的表型變異有關,一些疾病也是由重復擴張引起。分析STR,特別是長STRs的變化是理解其個體變異性和導至其不穩定的機制的重要步驟。
檢測STR比較簡單,不用下軟件,直接在線操作。用MISA-web (http://webblast.ipk-gatersleben.de/misa/) 中的MIcroSAtellite identification tool可以通過在輸入字段中指定相應的登錄號來從NCBI數據庫中檢索序列。MISA-web支持兩種不同的輸出格式:專有的MISA輸出格式和通用GFF3。GFF3輸出格式有助于將MISA網頁搜索結果集成到下游分析中。
功能預測
現代測序技術產生越來越詳細的基因組變異數據。然而,鑒于許多疾病性狀復雜,是多基因聯合作用,將單個變體或突變基因與表型相關聯的常規方法已有其局限性。做功能基因預測的工具就比較多了。
第一個在線工具是MutationTaster (http://www.mutationtaster.org/)。這個免費的在線應用可以快速評估DNA序列改變的致病潛力。
MutationTaster集成了來自不同生物醫學數據庫的信息,并使用已建立的分析工具。分析包括進化保守,剪接位點變化,蛋白質特征的喪失和可能影響mRNA量的變化。然后通過一個貝葉斯分類器評估測試結果,該分析器預測疾病潛力,一般的查詢在0.3秒內就可完成。
第二個網站是SNPdryad(http://snps.ccbr.utoronto.ca:8080/SNPdryad/ ),可預測在人類蛋白質中氨基酸取代會發生的有害作用。而且在準確預測有害nsSNP方面,SNPdryad優于其他算法。

第三個是個數據集,dbNSFP(https://sites.google.com/site/jpopgen/dbNSFP),為人類非同義單核苷酸變體(nsSNV)和剪接位點變體(ssSNV)的功能預測和注釋提供一站式資源,還有從外顯子中發現的大量SNV篩選和測序研究。dbNSFP創建了基于人類參考序列的所有潛在nsSNV和ssSNV的列表,并為每個SNV編譯了功能預測和注釋。
目前該數據集中包括82,832,027 個nsSNV和ssSNV,附加的數據庫dbscSNV編譯所有潛在人類SNV及其有害性預測,另外增加了的15,030,459個潛在功能SNV。
驅動突變
區分驅動突變與偶發突變對于了解致癌分子機制,以及鑒定預后和尋找治療靶標至關重要。MutSig可以分析在DNA測序中發現的突變列表,以便識別基因突變是驅動突變還是偶發突變。該軟件最初是用于分析體細胞突變,但也可用于分析種系突變。MutSig建立了在腫瘤形成期間突變過程的模型,分析每個基因,以鑒定比預期突變更頻繁的基因。
變異注釋
最新的測序儀器與SNP發現工具結合可用于識別大量可能的SNP,而在識別后的下一個問題就是注釋和選擇功能重要的SNP。這項任務也能用dbNSFP完成,不過更推薦用ANNOVAR。它可以利用不斷更新的信息來注釋不同基因組(包括人類基因組hg18,hg19,hg38以及小鼠,蠕蟲,蠅,酵母和許多其他基因組)檢測到的遺傳變異相關功能。ANNOVAR可以在一天內處理數百種人類基因組。
另外還有一個在線工具GeneTalk(http://www.gene-talk.de/),GeneTalk為分析人類序列變異的遺傳學家提供了直觀的基于網絡的界面。它幫助臨床遺傳學家,尋找有關特定序列變異的信息,并將該用戶與研究相同序列變異的其他用戶連接起來。

數據可視化
基因組瀏覽器不僅用于顯示最終結果,還能用于改進分析方法,測試數據質量和生成結果草圖。有助于創建和利用基因組瀏覽器新進展來改進分析結果并支持基因組數據的快速可視化。
ZENBU這個數據集,通過數據處理和交互式鏈接實現可視化數據挖掘,使用戶可以用BAM或制表符分隔(BED,GFF)序列對齊數據。

當然,除了這個在線工具外,前面介紹的IGV也能完成這個工作。
至此,一個完整WGS測序也就完成了。在臨床研究中,目前火熱的“精準醫療”和“個體化醫學”都需要準確的基因組參考序列,來幫助真正“精準”的基因組數據分析。WGS測序不論是在科研還是臨床診療上,今后都大有作為。
記者21日從國家乳業技術創新中心獲悉,該中心技術研發團隊成功研制出奶牛種用胚胎基因組遺傳評估芯片和“高產、抗病、長生產期”功能強化基因組預測芯片。該系列基因芯片具有完全自主知識產權,填補了我國基因芯片......
英國研究人員在新一期美國《神經腫瘤學雜志》上發表論文介紹,他們新開發出一種超快速腦腫瘤基因診斷方法,可以將腦腫瘤的診斷時間從此前的6至8周縮短至2小時。這種腦腫瘤基因診斷方法由英國諾丁漢大學等機構的研......
由新加坡科技研究局基因組研究所領導的科學家團隊,發布了迄今全球最大、最全面的長讀長RNA測序數據集之一——新加坡納米孔表達數據集(SG-NEx)。這一成果有望解決疾病研究中長期存在的技術瓶頸,使研究人......
經過20多年的努力,科研人員成功地對6種現存猿類的基因組進行了完整測序,為研究人類進化提供了近距離視角,這被英國《自然》雜志稱為“遺傳學的一個里程碑”。123名來自多個國家和地區的科研人員組成的團隊9......
昆明醫科大學第二附屬醫院2025年05月(至)2025年06月政府采購意向為便于供應商及時了解政府采購信息,根據《財政部關于開展政府采購意向公開工作的通知》(財庫〔2020〕10號)等有關規定,現將昆......
事件焦點:為什么是它?3月4日,中國將美國基因測序公司因美納(Illumina)列入“不可靠實體清單”,引發行業震動。這家生產基因測序儀的公司看似低調,卻是全球生物實驗室的“水電煤”——它的設備幾乎壟......
通過轉錄組測序獲得人的基因表達譜數據,能進一步挖掘疾病相關的生物標志物,為臨床診斷提供依據。目前,由于轉錄組測序無法溯源,導致不同實驗室及測序平臺產出的數據可比性和測序結果的準確性面臨挑戰。中國計量科......
14日,記者從四川農業大學獲悉,該校玉米研究所蘭海教授團隊利用全基因組選擇技術構建了西南玉米區域新的雜優模式,并運用該技術在國內首次選育出玉米新品種。相關成果近日發表在《植物科學前沿》上。蘭海介紹,該......
科技日報北京8月15日電(記者張佳欣)據最新一期《自然》雜志報道,德國康斯坦茨大學和維爾茨堡大學領導的國際研究團隊,對動物界最大基因組的擁有者——肺魚進行了基因組測序。肺魚基因組約為人類基因組大小的3......
據最新一期《自然·遺傳學》雜志報道,由多個機構組成的國際“端粒對端粒(T2T)”聯盟正在推進“反芻動物端粒-端粒”項目,旨在對300多種反芻動物的基因組進行測序。研究團隊期望通過測序得到的基因組圖譜,......